

About Hyper IgD Syndrome (HIDS), one of the Mevalonate Kinase Deficiency (MKD) illnesses.
 Hyper-IgD syndrome (HIDS), or HIDS disease, is caused by an inherited autosomal recessive gene mutation of the mevalonate kinase gene (MVK). Most patients have two mutations on the MVK gene, one from each parent to cause HIDS disease symptoms. However, there are some patients with only one identified mutation on the MVK gene, and some cases where no MVK mutations were found that have been identified as having clinical HIDS.
Hyper-IgD syndrome (HIDS), or HIDS disease, is caused by an inherited autosomal recessive gene mutation of the mevalonate kinase gene (MVK). Most patients have two mutations on the MVK gene, one from each parent to cause HIDS disease symptoms. However, there are some patients with only one identified mutation on the MVK gene, and some cases where no MVK mutations were found that have been identified as having clinical HIDS.
A clinical diagnosis can, and should be made after a full evaluation of symptoms, labs, and genetic testing. Almost all autoinflammatory diseases that are associated with genetic mutations also have cases that are “mutation negative” that have full symptoms and characteristics of the disease. New genetic testing methods have led to finding genetic mutations in previously “mutation negative” patients with clinical diagnosis of CAPS, and it is very likely that with more research, new genetic findings will be discovered for other autoinflammatory diseases.
This mutation causes a malfunction in the innate immune system’s inflammatory processes and a variety of symptoms. Like other recurrent fever syndromes, HIDS disease symptoms are caused by the patient’s genetic mutations and are not contagious at any time.
For a short overview of HIDS, click here.
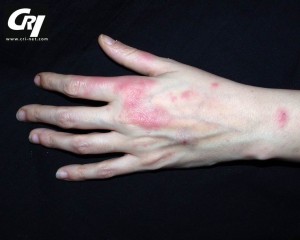
HIDS rash image from Le Clube Rheumatismes et Inflammation
Hyper-IgD Syndrome Symptoms
Symptoms are usually present before the age of one. However, because these symptoms, such as a high fever, are typical in early childhood, many doctors and parents may not recognize the symptoms as being abnormal until the child is older. A few cases of adult or later-onset HIDS have been reported.
Symptoms do vary greatly among individuals and even from flare to flare in the same person. The symptoms can also be progressive, as many parents report that their child develops more symptoms and symptoms may become more severe or pronounced with each flare over time. In some adults, the symptoms may become less frequent and less severe. But this does not happen in all cases.
Severity of symptoms, and signs of chronic inflammation, can vary greatly between each case of MKD, but these are generally lifelong diseases for most patients. Most patients with the HIDS form of MKD have significant symptoms during flares of their disease and can have some chronic inflammatory issues.
The most severe form of MKD is called mevalonate aciduria (MA), which involves a high level of chronic inflammation from birth leading to permanent and debilitating symptoms. More information about MA features and symptoms is on our comparative chart.
Common Hyper-IgD Syndrome Symptoms
High fever, often reaching 104.0 F or higher, that lasts less than 14 days is the main symptom. Fever flares occur usually every 1 to 12 weeks in childhood. The fever usually lasts 3 to 7 days, but a range of 2 to 12 days has been reported. Some patients report regular, almost predictable fevers, while in others the frequency of fevers is erratic and unpredictable. Some patients also report having feverless flares between fever flares. Feverless flares may have all or some of the symptoms of a fever flare, but may be milder and the fever does not develop.
With each flare, there are usually several other common symptoms that can include:
- Abdominal pain which can look like appendicitis to the observer
- Diarrhea (may have mucus and/or blood)
- Vomiting
- Mouth and/or genital ulcers
- Sore throat (pharyngitis)
- Joint pain, swelling, and redness
- Muscle pain
- Variable rashes
- Swelling of cervical lymph nodes
- Headache
- Enlarged spleen (Splenomegaly)
- Cutaneous vasculitis
- Cold chills
- Fatigue
Less Common Symptoms
- Conjunctivitis
- Enlarged liver (Hepatomegaly)
- Serositis
Rare Symptoms and Complications
- Amyloidosis has been seen in less than 10% of HIDS cases.
- Henoch-Schonlein purpura (HSP)
- Macrophage activation syndrome (MAS)
Other Conditions, Complications, and/or Symptoms Noted in Studies:
- Normocytic anemia
- Enlarged mesenteric lymph nodes
- Palpebral edema (swelling of the eyelid)
- Coronary arteritis
- Nephritis
- Diabetes insipidus
- Respiratory infetions
- Renal angiomyolipoma
- Cholestatic liver disease
- Colitis
- Retinitis pigmentosa
- Risk for pneumococcal infections
Flare Triggers
Vaccines, travel, illness, injury, birthday parties, big celebrations, excursions, school pressures, and any other stress, good or bad, to the immune system can cause a flare. In teens and adult women, menstruation is often a trigger for flares. Most with HIDS will have a flare within 24 hours of immunization. Some patients with HIDS have more frequent and severe flares in the winter and fewer and less severe flares in the summer. Sometimes the flares may seem random and the trigger can’t be identified.
Abnormal Lab Tests
Several tests are needed to help lead to a HIDS diagnosis and monitor a patient with Hyper-IgD disease.
IgD
The high IgD levels common to HIDS gave the disease its name of hyperimmunoglobulinemia D with periodic fever syndrome. However the test results, whether normal or high, are not conclusive.
Having a high IgD test result and recurrent fevers does rule HIDS in as a possible diagnosis, however is not diagnostic to HIDS. Other periodic fever syndromes can sometimes have elevated IgD, including FMF, TRAPS, and PFAPA. Also, around 20% of cases with confirmed genetic HIDS do not have high IgD or develop it later in life. Rarely do children under the age of 3 years have high IgD. Therefore having a normal IgD test result does not rule out HIDS as a possible diagnosis.
IgD levels do not correlate with disease severity. IgD cannot be used by itself to diagnose HIDS. Read more about IgD in fever syndromes here.
IgA
80% of patients may also have a high IgA test result, often in correlation with elevated IgD.
ESR and CRP
The inflammatory tests ESR and CRP are often high during a flare and normal when not in a flare.
White Blood Counts
Neutrophils, and/or monocytes, and/or the overall WBC are also often high during a flare. This often leads to unnecessary treatment with antibiotics or a misdiagnosis as a recurrent virus or other infection.
In some, IgD, ESR, CRP, and/or WBCs may be high when not in a flare as well.
Mevalonic Acid Urine Test
MKD causes high levels of mevalonic acid in the urine during a flare. However, this is a difficult test to perform and not always a reliable diagnostic test. Patients with genetic confirmed HIDS have been known to have normal mevalonic acid tests. A 2014 study presented at the European Pediatric Rheumatology Congress showed that patients without MVK mutations can have elevated mevalonic acid in their urine. Having a normal test does not rule out HIDS disease as a possible diagnosis. This test is often reserved for research purposes.
Diagnosing Hyper-IgD Syndrome
All of the tests above are helpful in determining if a person has a periodic fever syndrome that could be HIDS, but each of these tests alone do not diagnose HIDS specifically.
The only way to positively confirm a genetic HIDS diagnosis is through genetic testing.
It’s ideal to have a full panel of genetics testing for similar hereditary autoinflammatory diseases if HIDS disease is suspected. This not just to test for HIDS, but to also rule out other periodic fever syndromes, such as FMF, CAPS, and TRAPS, which have many symptoms and abnormal lab results similar to HIDS. Here is a list of some of the genetic fever panels available in the U.S.
If no mutations are found after genetic testing, then a clinical diagnosis of HIDS can be made based on symptoms, laboratory tests, and negative genetics for similar conditions, such as FMF, CAPS, and TRAPS. In clinical HIDS, the recommended treatments are generally the same as for genetic cases of HIDS.
Inheritance
HIDS is considered a recessive disorder. This means that you need to inherit one mutation from each parent to have the symptoms of HIDS. However, some patients with only one known HIDS mutation show symptoms of disease, even when their parent does not. It’s not completely understood why these patients have symptoms with only one mutation; modifier genes are a current research interest that could explain this. Studies have shown that symptomatic patients with a single mutation on the MVK gene, can have the same symptoms and severity of disease as those with two HIDS mutations.
There is also a recognized variant HIDS form of this disease in which patients have the symptoms of HIDS but no mutation is found.
HIDS is most common in those with a Dutch or French ancestry, but has been reported in a range of ethnic groups.
Most Common Hyper-IgD Syndrome Treatment Options are Experimental and “Off-Label”
HIDS does not have a single treatment that works well for every patient. Currently only Ilaris (canakinumab) has FDA approval for HIDS. For HIDS treatments, we also refer to research studies in our discussion about the use of “off-label” treatments that were used in controlled clinical trials. “Off-label” means that the drug does not currently have FDA approval for use as a treatment for HIDS, but are approved for other diseases. Most autoinflammatory diseases do not have FDA-approved medications, so prescribed medications to treat these diseases are written “off-label.” Experimental use of medications, based on clinical research studies, is very common for rare diseases.
There are also no known treatments that effectively prevent all HIDS flares in most patients, but most with HIDS improve greatly once on proper treatment.
Most medications prescribed for HIDS at this time are experimental, and patients need to work with their doctor in what “off-label” medications to try for their disease. We are merely presenting a discussion of what has been prescribed during clinical research, and are not suggesting, or recommending the use of one medication over another for use with HIDS.
Most Effective HIDS Treatment Options Include:
Most patients are first prescribed treatment with ibuprofen and acetaminophen (over the counter non-steroidal anti-inflammatory drugs-aka NSAIDS) to control the fevers. This is usually before a HIDS diagnosis is made. However NSAIDS only help to treat symptoms and do not block the inflammation causing the flares. Most patients cannot control flares with NSAIDS. After NSAIDS, the most commonly prescribed medication for HIDS is oral prednisone, at a dose of 1mg/kg/day reports the authors of The Autoinflammatory Diseases, to be given at the first sign of a flare. Prednisone has been found to help patients more effectively if administered at the first onset of symptoms. Although, prednisone in some patients has been found to cause flares to occur more frequently.
Il-1 inhibitors anakinra and canakinumab have been found to be the most effective treatments for HIDS, although not all patients have a complete response to these treatments. HIDS researchers recommend that after prednisone, Il-1 inhibitors are the first biological treatments to try in HIDS.
In some, Kineret has been prescribed “off-label” for daily use or for “on-demand use.” For patients with less frequent flares, doctors may often prescribe off-label Kineret on-demand for use during flares only. Some doctors prescribe Kineret off-label for daily injections for patients with more frequent HIDS flares or who are more symptomatic in an attempt to reduce the frequency of flares and symptom severity.
In the study entitled Anakinra for the Treatment of Hyper-IgD with Periodic Fever Syndrome in Children, researchers report for HIDS patients, “Dosage (of Kineret) most often was initiated at 1mg/kg with further increases when needed up to 3–5mg/kg/day.” This lead to, “The majority (8 [72%] ) of 11 children with HIDS demonstrated a beneficial response to anakinra therapy…”
Several studies have also shown canakinumab (Ilaris) to be an effective treatment for HIDS and it is now FDA approved for HIDS treatment. According to Juan Ignacio Arostegui, MD, PhD in a 2013 study, “CAN (canakinumab) in active HIDS markedly reduced the rate of acute flares, and a sustained good/excellent disease control was observed.” In this study, most patients were given a dose of 4mg/kg every 6 weeks. With this the median flares went from 5 in 6 months to 0 in 6 six months.
According to 2015 Hyper-IgD syndrome/mevalonate kinase deficiency: what is new?, one patient was treated with 4mg/kgof canakinumab every 4 weeks and had complete clinical response at that dose.
Other Treatments
Several other medications have been tried experimentally, and again “off-label,” with HIDS patients with varied success. These include Enbrel, statin drugs, daily NSAIDS, and colchicine. Studies have shown that a few patients have found relief for HIDS symptoms with Enbrel. However, it is recommended that only if a patient fails the Il-1 biologics, Kineret and Ilaris, to then try a TNF-inhibitor such as Enbrel or Humira. Dr. Simon reports, “Anti-TNF therapy might be effective in MKD, but the effect is mostly partial and therapy failure and clinical deterioration have been described frequently in patients on infliximab or etanercept.”
Simvastatin was studied as a treatment in some adults with HIDS, but it did not effectively stop flares and statins are generally not the preferred treatment for adults or kids with HIDS. Comprehensive studies have not been done with Simvastatin in children with HIDS, and statin drugs are not generally recommended by researchers as a treatment for children with HIDS.
Colchicine has not proven effective in helping to control HIDS fevers and flares report the authors of Hyperimmunoglobulinemia D (hyperIgD) and periodic fever, but some patients have found that colchicine has helped to relieve joint and muscle pain. Some have also found that combining colchicine with an Il-1 inhibitor, such as Kineret or Ilaris, can also help better control symptoms.
As of 2014, there are a two case reports of tocilizumab (Actemra), which is an interleukin-6 inhibitor, being used to treat HIDS. In one study, it was considered a successful treatment in a 13-year-old girl after several other treatments had failed. In another study from the National Amyloidosis Center in London, U.K., it was reported that, “All patients reported improvement in symptoms.” This study included one HIDS patient.
Long Term Prognosis of HIDS
It was once thought, and is still often stated, that HIDS is something children will outgrow. However, since the discovery of the HIDS gene and mutations, and the identification of more adult cases, it is now known that most adults do suffer from some degree of HIDS symptoms, ranging from mild to more severe. What studies have shown is that some, but not all, HIDS patients may suffer from fewer and/or less severe flares as adults than they did as children.
The following statements come from a study that included 103 HIDS patients published in the journal Medicine in 2008 by the International HIDS Study Group:
HIDS is a severe disease that starts early in life with lifelong recurrent attacks of fever accompanied by a variety of symptoms, including lymphadenopathy, abdominal pain, arthralgia, vomiting and diarrhea, skin lesions, and aphthous ulcers.
There was a significant decrease in frequency of attacks with increasing age, although no patients had a remission.
After the age of 20 years, 17.8% of patients continued to have attacks more than 12 times per year, while 50% of patients still had more than 6 attacks per year. Thirty-three of 45 patients above the age of 20 years had fewer attacks after the age of 20 years than in the first decade of life.
HIDS is a lifelong chronic condition that can be managed with successful treatment. For some adults, managing their disease is easier than it was when they were a child. It’s not known why some adults improve, but in some cases it may be due to improved understanding and treatment of their condition.
Rarity of HIDS
HIDS is one of the rarer autoinflammatory conditions. As of 2014, estimates show there are about 300 total confirmed HIDS cases in the world. Most are found in The Netherlands. Less than 100 HIDS patients have been identified by researchers in the U.S. Likely this is an under-diagnosed autoinflammatory disease, in part because the symptoms can very closely resemble PFAPA, another recurrent fever syndrome.
References
- GeneDx: Periodic Fever Syndromes Panel (7 Genes)
- American College of Rheumatology: The Clinical Significance of a Single MVK Mutation in HIDS
- Clinical Rheumatology: Different clinical presentation of the hyperimmunoglobulin D syndrome (HIDS) (four cases from Turkey)
- Rheumatology International: Incidence and clinical features of hyperimmunoglobulinemia D and periodic fever syndrome (HIDS) and spectrum of mevalonate kinase (MVK) mutations in German children
- Mediators of Inflammation: Biological Treatments: New Weapons in the Management of Monogenic Autoinflammatory Disorders
- van der Hilst JCH, Bodar EJ, Barron KS, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine. 2008;87(6):301–310.
- Case Reports in Rheumatology: A case of hyperimmunoglobulinemia d syndrome successfully treated with canakinumab
- Italian Journal of Pediatrics: Autoinflammatory syndromes: diagnosis and management
- DermNet NZ: Hyperimmunoglobulinaemia D with periodic fever syndrome
- Firestein: Kelley’s Textbook of Rheumatology, 8th ed.: Hyper-immunoglobulin D Syndrome
- HIDS.net: Treating the Disease
- HIDS.net: Life with a fever every ten days
- Hereditary Autoinflammatory Syndromes with Emphasis on Hyper-IgD and Periodic Fever Syndrome; Dr. Anna Simon
- ISSAID 2013: Single MVK Mutation and Recurrent Fevers
- Pediatric Rheumatology: Tocilizumab in autoinflammation and AA amyloidosis
- NIH: Hyper-Immunoglobulin A in the Hyperimmunoglobulinemia D Syndrome
- NIH: Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome.
- Johns Hopkins Arthritis Center: The Expanding Spectrum of Systemic Autoinflammatory Diseases: Misadventures in the Genomics of Inflammation by Dan Kastner, MD, PhD, Clinical Director, NIAMS, Chief, Genetics and Genomics Laboratory, NIAMS/NIH/DHHS
- Rheumatology: Efficacy of interleukin-1-targeting drugs in mevalonate kinase deficiency
- Rheumatology: Canakinumab Treatment In Active Hyper-IgD With Periodic Fever Syndrome
- Blackwell Publishing: Anakinra for the Treatment of Hyper-IgD with Periodic Fever Syndrome in Children
- Orpha.net: Hyperimmunoglobulinemia D (hyperIgD) and periodic
- PRINTO: Mevalonate Kinase Deficiency (MKD) (or Hyper IgD Syndrome)
- Autoinflammation: HIDS
- Pediatric Rheumatology: Diagnostic value of urinary mevalonic acid excretion in mevalonate kinase deficiency (MKD)
- Research Gate: Different presentations of mevalonate kinase deficiency; a case series
- Springer Link: Hyper-IgD syndrome/mevalonate kinase deficiency: what is new?
{kind=link}